






























 Arguments
Arguments


















Not pHraud but pHoolishness
Posted on 5 January 2015 by Guest Author
This is a re-post from Richard Telford's blog, Musings on Quantitative Palaeoecology
By a curious coincidence, many climate sceptics are also ocean acidification sceptics. Some, for whom a rose by any other name would not smell so sweet, try to hide their rejection of reality behind semantics, arguing that ocean acidification should be called ocean neutralisation or ocean dealkalinisation. Others try to disprove ocean acidification with misremembered school chemistry, and yet others use dubious statistics.
There is an outbreak of the latter at WUWT, where Mike Wallace presents an analysis of ocean pH data that the ever gullible Anthony Watts finds “compelling”.
Wallace appears to have taken an objection to this figure by Richard Feely which shows atmospheric CO2 concentrations measured at Mauna Loa and ocean CO2 concentrations and pH measured in the nearby ocean.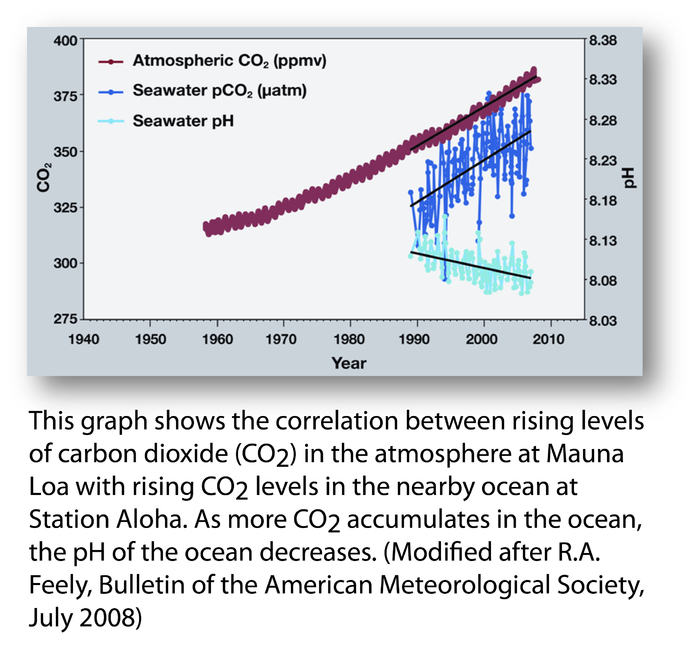
Wallace’s complaint is that this figure omits 80 years of data on ocean pH and that this omission represents “pHraud” that “eclipses even the so-called climategate event.”
Wallace proceeds to compile all available ocean pH data from the World Ocean Atlas and calculate annual mean pH over the last century.

Wallace’s naïve analysis
A global-ocean mean pH: what could possibly go wrong?
Consider what would happen if one simply took all available temperature data used this to estimate annual mean temperatures over the last 100 years, rather than calculating anomalies and gridding quality checked data. The result would obviously be nonsense. Changing geographical and seasonal biases in data availability, and incorrect data would corrupt the analysis. Wallace’s analysis suffers from exactly the same problems.
Geographical variability in ocean pH is large. Upwelling area have the lowest pH as the water upwelling from the deep oceans has high CO2 concentrations from decomposition of sinking organic matter. The geographical coverage of ocean pH measurement is extremely unlikely to have remained constant over the instrumental period. Any analysis that fails to take this into consideration is doomed.

Geographic variability in ocean pH. Apologies for the rainbow scale.
Intra-annual variability in pH is also high. Intense photosynthesis during algal blooms can raise pH, and seasonal upwelling can lower it. If the seasonal coverage of ocean pH measurements has not remained constant over time, biases will result.
The data Wallace analysed are easily downloaded from the World Ocean Database, and the metadata examined for geographic or seasonal patterns in data availability. I’ve analysed the data on a per-cast basis. Each cast collects data from several different depths.

Location of casts by decade
Geographic patterns of data availability vary from decade to decade (and even more on an annual basis).

Season of northern extra-tropical casts by decade
The seasonal pattern of data availability in the northern extra-tropics (which represent the bulk of the data) also vary over time.
The changing geographic and seasonal patterns in data availability means that simply calculating the mean pH for each year will give all sorts of spurious trends in the analysis. Even gridding the pH data would be difficult. They are probably best used to validate model output.
Certainly, Wallace’s “compelling” analysis is junk. I hope the rest of his PhD is better than this pHoolishess.








Even without knowing the means whereby "20th Century Ocean pH" was derived, Wallace's graph immediately raises concern in that the pH values he gives vary widely on an annual basis before about 1975, whereas thereafter that variation is small. This is reminiscent of claims of large variability of global atmospheric CO2 prior to 1959, for example by Beck.In both cases, the huge changes in concentration claimed from one year to the next without any plausible mechanism to cause them should immediately suggest the falseness of the reported data.
This nonsense by Wallace reminds me of the CO2 rubbish of Ernst Beck. I suspect that Wallace will follow Beck and publish in E & E.
I'm curious how Wallace proposes that atmospheric concentrations of CO2 have been rising and yet ocean is not becoming more acidic.
If the data are so dodgy why did NOAA put them on its website without warning that the data were unreliable?
Nice try, but NOAA isn't culpable for Mike Wallaces' ignorance.
3. Rob Honeycutt
Perhaps Wallace has been fooled by the sort of nonsense that Tom Segalstad writes here:
https://docs.google.com/file/d/0B74u5vgGLaWoM0FaOUxrZ21FSmM/edit?pli=1
where he claims that, ""the upper 200m of ocean water contains enough dissolved calcium to bind all anthropogenic CO2 as precipitated calcium carbonate ... without effecting the ocean's pH" (p.818)
Segalstad is wrong.
The overall equation for the reaction of CO2 with calcium ions to produce solid calcium carbonate is as follows:
CO2 + Ca++(aq) + H2O ==> Ca++Co3--(s) + 2H+(aq)
The reaction produces hydrogen ions (2H+aq). You CANNOT precipitate solid calcium carbonate by combining aqueous CO2 and calcium ions WITHOUT at the same time producing hydrogen ions, and thereby reducing pH. (Of course, the H+(aq) + OH-(aq) <==> H2O ( K = c.10^-14 ) equilibrium is simultaneously maintained, but there is still an overall increase in hydrogen ion concentration)
Hmm. Does this mean that the deniers are moving away from their inaccurate claims about temperatures and over to other topics. Wonder why.
William:
The CO2 data for the oceans are not unreliable, just very incomplete. NOAA put them out because they are valid measurements. The measurements have been made well at least since the 60's and probably longer. The problem is that there is an attempt to create a global record by simple averaging of available data--this is not a smart way to go about producing an average where pH can vary by large amounts depending on the strength of upwelling. I
I guess I get irritated with people that are opposed to funding the monitoring effort but then claim that we cannot determine the trend. If one thinks that there is no trend, there should be more funding to show that, not less. Since 2000, funding for environmental science at the US National Science Foundation for environmental science has dropped by about 20%.
William, try reading for comprehension: the older data are not dodgy, the data are simply incomplete, plus dissolved CO2 concentration is not spacially or temporaly homogeneous. Not taking those facts into account is what is dodgy.
Jim Eager @ 9 fair enough but dodgy or incomplete or not spatially or temporally homogeneous or whatever, I am surprised Feely and Sabine made no reference to these data even if only to explain why they chose not to use them. I think they should have done and had they done so, none of this would have eventuated thus denying sceptical blogs another so called "example of sharp practice" with which to regale their readers.
And as an aside, a more recent paper published in PLoS One in 2011 using direct measurements of ocean pH, has shown marine species that are regularly exposed to much greater changes in pH than those forecast for the future and that some species currently are exposed to pH levels more acidic than those forecast for 2100 (http://tinyurl.com/6t9fjly} The paper is also summarised at https://scripps.ucsd.edu/news/1875 From this it may be that ocean acidification may be less deleterious than currently believed.
william - Apart from the 'blaming the victim' issue of putting the onus of pseudo-skeptic misunderstanding on the scientists, Feely and Sabine did tell Wallace about the issues with data coverage; he apparently followed by impugning their science and motives. Not IMO honorable behavior on Wallaces part.
The data coverage issues of past ocean sampling are well known to those familiar with the science - hence there is little reason to repeat common knowledge in the field in every paper.
This is why we have peer-review. The data is made available for the use of scientists with the appropriate domain-knowledge and expertise to be able to use it. This does of course also mean that amateurs with an agenda can use it. If Wallace had tried to publish however, expert reviewers would have immediately pointed out the issues to him. But then I doubt Wallace is really interested in ocean pH, only in ammunition to support a viewpoint based on political values.
Trying to blame Wallaces' ignorance of rather basic science on Feely and Sabine is rather amusing though.
"From this it may be that ocean acidification may be less deleterious than currently believed"
That's just wishful thinking. The near-collapse of the US North Pacific oyster fishery due to the die-off of larval oysters from too corrosive seawater highlights the rather obvious flaw in your reasoning. And when we look at the geological record, many marine calcifiers went extinct in the past when the oceans acidified at rates much slower than present-day.
The one of the greatest problems with most laboratory-based ocean acidification experiments is that they don't actually simulate conditions that marine organisms face throughout their lifecycle. That was the problem with the larval oysters - they are actually exposed to conditions much more corrosive than consideration of atmospheric CO2 alone would suggest, and their early lifestages are a time of great vulnerability to carbonate saturation state (corrsiveness of seawater).
So quantification of actual conditions that marine organisms face is essential if one wants to accurately forecast how they might respond to ocean acidification. The Hofman paper is right about that.
slioch @6: Your eqn wrong. Calcification is a SOURCE not a sink for CO2.
Better to use our eqns 1 and 4 from the OA not OK series. (Click 'OA not OK' button to left).
Rob Painting I must have misunderstood the paper but I certainly got the impression that the authors had looked at the conditions to which the various species were exposed and at the variables in pH conditions to which these species were exposed. These variables encompassed pH values more acid than those considered likely to be extant in 2100. This is what the Scripps Institute of Oceanography said about the paper "In some of their study areas, they found that the decrease in seawater pH being caused by greenhouse gas emissions is still within the bounds of natural pH fluctuation. Some areas already experience daily acidity levels that scientists had expected would only be reached at the end of the 21st Century". In the example you give the area was one in which upwelling of colder more acidic sea water occurs. Perhaps this also had an impact. I'm not trying to blame Feely and Sabine for anything Wallace did or said although their attitude as reported doesn't seem particularly pleasant. Still I don't know what Wallace's attitude to them was like. They may have been hacked off with his approach.
"These variables encompassed pH values more acid than those considered likely to be extant in 2100"
Yes, I don't think this has been communicated particularly well by the scientific community.
There is no point comparing say the upwelling region of the California Current system with the projected average global pH and saturation state in 2100 because local marine organisms may be periodically exposed to conditions exceeding those right now. What the experiment should be simulating are projected local future conditions. In some instances this may be equivalent to atmospheric concentrations exceeding 3000 ppm.
Clearly there are serious limitations to lab experiments because marine life in the real ocean generally doesn't have ocean pH and carbonate saturation state suddenly ramped up to maximum volume. But on the other hand, exposing only adult populations, which are typically less vulnerable, isn't realistic either. That's where studies of naturally acidified marine environments are useful. With a few exceptions, most marine calcifiers typically don't fare too well.
14. Doug Mackie
Your 'OA not OK' button does not appear to work, so I have no idea to what equations you refer, but it is certainly not correct to state that "calcification" (if by that you mean the conversion of dissolved calcium ions to solid calcium carbonate) is a source of CO2. That is simply impossible.
If you can post the equations to which you refer I can answer your query.
Incidentally, there is an alternative (though equivalent, due to the H+ + OH- <=> H2O equilibrium) overall equation for the production of solid calcium carbonate that may be written thus:
Ca++ + CO2 + 2OH- ==> Ca++CO3-- + H2O
This also, of course, shows the process to involve absorption of CO2 (ie it is a sink for CO2), which is indubitably the case.
[KC] The button links to a list of posts on ocean acidification, rather than the articles directly - that's not very intuitive. Doug was referring to the first four posts (ignoring the introduction) from the bottom of that list. Here are the direct links for you:
1 July 2011 by Doug Mackie
5 July 2011 by Doug Mackie
8 July 2011 by Doug Mackie
10 July 2011 by Doug Mackie
Slioch @17:
1) Rather than using the OA not OK button, which just leads to the results of a search, try Part 1 and Part 2 of the summary of the OA not OK series, following further links for the more detailed discussion.
2) From Part 1, we can link through to the first post of the series which shows the following equation:
That shows calcification, as a reaction between Ca2+ and 2HCO3- is clearly a source of CO2 rather than a sink. Part 1 of the series also contains the crucial advise that:
Taking that advise, we can note that while you can write alternative equations that balance, that does not show that they are the preffered reaction (ie reaction spontaneiously occuring at the greatest rate) in given conditions. If the reaction is biological mediated, the equilibrium constraints which you ignore become even more stringent in that biologically mediated reactions often have only one pathway.
3) From the summary of part 5, we also learn that:
That is, 91% of inorganic carbon is in the form found in equation 1, giving a strong reason why that is the preffered reaction and your equations show very slow subsidiary reactions if that. That your equations require the simultaneious interaction of three molecules would further lower the rates unless there are reasonably stable intermediates (in which case please break the reactions apart to show the intermediate steps).
Thanks to the moderator for providing the links to the 'OA not OK' articles.
There is nothing therein that contradicts my previous posts and, once again, the assertion that calcification is a source of CO2 rather than a sink is simply wrong.
Tom Curtis (@18) has helpfully posted the relevant equation from OA not OK above, but, I'm sorry, Tom, you clearly do not understand what you are writing about.
I posted the OVERALL equation (neither you nor I have shown the mechanism of the numerous reaction steps , nor is that necessary) with respect to the precipitation of solid calcium carbonate from an aqueous solution containing calcium ions (you will note that in in both of my previous posts on this subject I referred to an "overall" equation). The overall equation provides a summary of the overall changes in constituents in that process, and, as I have stated previously, indubitably shows that the process consumes CO2 (and reduces pH).
The equation that you show, from the 'OA not OK' site, is an INTERMEDIATE equation from which, on its own, no conclusion can be drawn about OVERALL changes. (The 'OA not OK' site's assertion that calcification produces CO2 is simply wrong)
Hopefully this should become clear from the following:
The question is: "does the precipitation of solid calcium carbonate from a solution containing calcium ions a) cause an absorption of CO2 and b) cause a reduction in pH (ie an increase in hydrogen ions). The following shows that the answer to both questions is "YES".
Your equation shows bicarbonate ions being consumed. What is the source of those bicarbonate ions? They come originally from gaseous CO2 dissolving in water. The intermediate steps are as follows: (note: I will write all these equations as 'one way' equations, though they are in fact equilibria, since I am considering the process leading to precipitation of the product CaCO3).
Equ1. 2CO2(g) ==> 2CO2(aq)
That CO2(aq) then produces bicarbonate ions:
Equ.2 2CO2(aq) + 2 H2O ==> 2HCO3- + 2H+
(I've doubled those equations since we need 2HCO3- below)
If those bicarbonate (HCO3-) ions then react with calcium ions, then it as shown in the equation that you (and 'OA not OK') post:
Equ3. Ca++ + 2HCO3- ==> CaCO3(s) + CO2 + H2O
If we then ADD those three equations together, we get the overall equation:
2CO2(g) + 2CO2(aq) + 2 H2O + Ca++ + 2HCO3- ==> 2CO2(aq) + 2HCO3- + 2H+ + 2HCO3- + 2H+ + CaCO3(s) + CO2 + H2O
Cancelling leads to the overall equation:
CO2(g) + H2O + Ca++ ==> CaCO3(s) + 2H+
which is what I gave in the first place (@6) and which shows that CO2 is absorbed and hydrogen ions produced (lowering pH) in the process.
I hope that is now clear.
[Rob P] - Calcification is a source of CO2 - as Doug Mackie has already pointed out. It seems a few people with chemistry backgrounds get this wrong, so you have plenty of company. There is a great deal of scientific literature on this. For example see the Royal Society Report on Ocean Acidification (2005):
"The formation of CaCO3 leads to an increased CO2 concentration in the water. This apparently counterintuitive behaviour arises because two ions of bicarbonate (HCO3 – ) react with one ion of doubly charged calcium (Ca2+) to form one molecule of CaCO3, which leads to the release of one molecule of CO2. Some of this released CO2 is converted to bicarbonate by the buffering process, outlined above and in Annex 1. Under current conditions, for each molecule of CO2 produced during calcification about 0.6 molecules are released, potentially to the atmosphere, while the rest is taken up by the bicarbonate-carbonate buffer (Ware et al 1992)"
And that would be this from the OA not OK series:
Sorry, I must have clicked the paste button twice. The three equations added together should have been:
2CO2(g) + 2CO2(aq) + 2 H2O + Ca++ + 2HCO3- ==> 2CO2(aq) + 2HCO3- + 2H+ + CaCO3(s) + CO2 + H2O
Slioch
The problem is that, while your equations are right in terms of stoichiometry, presenting the overall equation that way gives the incorrect impression that that increasing CO2 will lead to increasing calcium carbonate production. In fact the opposite will happen in the short term because increasing CO2 shifts the pH in a direction that shifts the H2CO3, HCO3, CO3 equlibrium away from CO3 and toward H2CO3. This will tend to make the conditions needed to form CaCO3 rarer in the ocean.
Typically the reaction involving calcium carbonate formation it is considered separately from the reaction involving hydration of CO2 to H2CO3 because the equlibrium concentration of HCO3 and CO3 in the ocean is largely determined by the base cation concentration (the alkalinity) and pH, while the equilibrium CO2 concentration, on the other hand, is largely determined by temperature and atmospheric CO2 concentrations.
Together, these two reactions allow the calcium carbonate pool in the ocean to act as a buffer for pH in the very long run. As CO2 rises, calcium carbonate dissolves, consuming protons and consuming CO2 in the process to make bicarbonate ions. As base cation concentrations increase or CO2 decreases, calcium carbonate is formed, producing protons and CO2.
Its a pair of reactions that are way too slow to keep up with changes due to anthropogenic emissions however.
Have the deniers missed an easy reuse of a broken trick? Eyeballing that Station Aloha graph from 2000 (or, if it were clearer, maybe even the magical 1998?), it's clear that there's been a hiatus(!) in the seawater pH decline. In fact, if you choose your points carefully, has it actually risen? ;o)
On a more serious note, assuming that the graph shows seawater pH for surface layers, can one presume that the change in the graph since about 2000, being reminiscent of changes in the temperature graphs, is similar in being caused by more CO2 being pushed to deeper levels?
Stephen @ 21
Take a look back at the origin of this discussion of ocean chemistry. It began at #6 when I used that equation to counter Tom Segalstad's false assertion that ""the upper 200m of ocean water contains enough dissolved calcium to bind all anthropogenic CO2 as precipitated calcium carbonate ... without effecting the ocean's pH". It seems to me that claiming that increased CO2 will not increase ocean acidity is an important falsehood to counter, and that equation is a simple way of so doing.
(for info. on Tom Segalstad see, for example, http://www.desmogblog.com/tom-segalstad )
The equation 1. presented in the OA not OK article does not obviously address that issue, and the subsequent discussion was on a separate issue.
Slioch,
Understood. But the equation you presented is not the actual reaction that would take place if you added CO2 to the ocean.
When CO2 dissolves in water at pH 8 it largely dissociates to bicarbonate ions and protons. The release of the protons actually causes equation 3 in your post @19 above to want to run the opposite direction than you have it, because the increased acidity shifts the carbonic acid-bicarbonate-carbonate equilibrium away from carbonate and toward bicarbonate and carbonic acid. So it should read..
1) H2O + CO2 + CaCO3 ==> Ca++ + 2HCO3-
If you add that to the equations involving hydration of aqueous CO2 to form carbonic acid and the subsequent dissociation of most of the carbon acid to bicarbonate given a pH~8, you get
2) 2H2O + CaCO3 + 2CO2 ==> H+ + 3HCO3- + Ca++
Actually, the reaction mostly occurs after the aqueous CO2 has equilibrated with bicarbonate, so...
3) 2H+ + CaCO3 + 2HCO3- ==> H+ + 3HCO3- + Ca++
they are stoichiometrically equivalent, but the net effect (after reequilibration) is closer to the latter under current pH. Calcite dissolution has the net effect of removing protons and, thus, lowering acidity. It also consumes CO2. Calcite formation does the opposite.
So, Segalstad is wrong on two counts. If the Ca was to bind to anthropogenic CO2, it would force reaction 1 to run backward, which would actually increase the CO2 in the oceans, not decrease it. This would perforce increase acidity after equilibration with bicarbonate. You are also essentially removing basic cations (Ca++) and alkalinity in the oceans and decreasing the equilibrium concentration of bicarbonate, and therefore the capacity of the ocean to absorb CO2 and store it as dissolved inorganic carbon.
Second, adding CO2 would actually tend to retard formation of calcium carbonate anyway. So it's pointless to say that enough dissolved Ca is present to bind the bicarbonate, because if anything calcium carbonate will be dissolving with more CO2, causing Ca++ concentrations to rise further in the future. You'd have to add an unimaginable amount of base to the ocean to do what Segalstad is suggesting. It's kind of like saying there is enough dissolved gold in ocean water to make everyone rich and then wondering why we aren't all rich.
What is true is that there is enough calcium carbonate in the ocean sediments to largely neutralize athropogenic CO2 dissolved in seawater, but that process is much slower than release of CO2 has been and will take thousands of years to complete.
I should mention that many people have the mistaken idea that to be consumed, CO2 must be converted to a particulate form, like a plant or rock. But, from the point of view of water chemistry and the interaction of the ocean with the atmosphere, the speciation of dissolved inorganic carbon is extremely important.
The vast majority of ocean carbon is as bicarbonate, so more dissolved inorganic carbon as bicarbonate in the ocean means less CO2 in the atmosphere, given a closed budget. Less inorganic carbon as bicarbonate means more CO2 in the atmosphere. By removing base cations, the precipitation of calcium carbonates reduces the amount of dissolved bicarbonate in the ocean, and thus reduces the total amount of CO2 the ocean can absorb.
Siloch: Stephen Baines is correct about the importance of speciation. See the speciation fig.3 (below) in part 8 of OA not OK series and the step through to make your own figure in Appendix 2 of the book we did (link someone?) .
To counter denialists it is important to be rigorous. I mean no disrespect but your simplified eqn @6 was misleading and incorrect. Why give them a chance to misunderstand and misinterpret?It is worth recalling that it is not 'acidity' per se that is the problem for CaCO3 dissolution. Instead the key point is that changing [H3O+] causes a change in [CO3=] and it is this change in concentration of carbonate that causes problems. Adding CO2 to atmosphere causes CO2 to enter ocean. This increases total dissolved inorganic carbon (DIC) in the ocean but decreases the fraction of DIC that is carbonate ions. (See parts 15 and 16 of the OA series).
[Rob P] - pic & link provided.
Thanks to Stephen and Doug for their posts. I have a friend arriving soon for several days and I know that once she's here thoughts about ocean acidity (or anything else for that matter) will recede from my mind, so I will have to study your posts and possibly come back much later. I've read the first four parts of OA not OK and it is all very straightforward, except for the subject in dispute (the answer to which, I suspect, may turn out to be a question of time scales), but I will have a look at the rest once I have the opportunity.
Meanwhile, if I may, could I pose a question:
Suppose we have an Earth in which oceans and atmosphere are more or less at equilibrium and in which atmospheric CO2 and oceanic dissolved inorganic carbon is not changing very much over the long term (in other words an Earth in which no great volcanic or mountain building activity is occuring and no naked ape is chucking fossil carbon into the atmosphere like there's no tomorrow). Into this unchanging world a new lifeform evolves that causes the deposition and sequestration of huge quantities of calcium carbonate on the ocean floors. What then happens to the concentration of CO2 in the atmosphere over time?
[Rob P] - A scenario similar to that which you propose most likely did happen in Earth's past. Given the information provided by Doug Mackie & Stephen Baines, what do you think would happen?
Slioch @28
I'm not a real chemical oceanographer, but I'll take a stab at this. Doug can correct the specifics later. I'd be interested to hear his opinion as I sometimes have to teach this stuff.
You have to think of the combined ability of both the ocean and the sediment to store carbon from the atmosphere when answering this question. By producing calcium carbonate, a calcifying organism is removing a Ca2+ ion from solution by bonding it to a carbonate ion. That has two effects.
First, it removes the Ca2+ ion from solution and places it in sediments. By reducing the base cation concentration in the ocean, this reduces the total amount of bicarbonate and carbonate ion in solution within the ocean at equilibrium. With respect to the atmospheric CO2, this transfer is stealing from Peter to pay Paul.
Second, calcification is not an efficient way to use Ca to store C away from the atmosphere, at least relative to having dissolved calcium ions. In calcium carbonate, you store one mole of C (as CO32-) per mole of bivalent Ca. When dissolved in the ocean at current pH, univalent bicarbonate (HCO3-) is the most abundant form of dissolved inorganic carbon at current pH. To maintain charge balance, two bicarbonate ions are in kept solution for every dissolved bivalent Ca ion.
So yes, the sudden appearance of a massively calcifying organism would increase atmospheric CO2 by moving removing more storage capacity from the ocean than it adds to the sediments. Therefore some of the huge reservoir of carbon stored as dissolved bicarbonate and carbonate in ocean water would be free to equilibrate with the atmospheric reservoir of CO2. The exact effect would vary a little depending on the pH, chemistry and temperature of the ocean. The effect becomes more neutral as you consider pHs above current levels because dissolved carbonate becomes more abundant.
Of course, it's a hypothetical example, as such an organism would find it increasingly difficult to calcify as the ocean pH became more acidic and carbonate became less abundant in ocean water. Also, we have not discussed the secondary effect of calcium carbonate production on storage of organic carbon. That is a different kettle of fish entirely.
@ Stephen & Doug et al
As I only did Chemistry in First Year (and that was a long, long time ago) I am struggling to keep up with the subtleties here. Would I be correct in thinking that this represents a good example of le Chatelier's Principle in action?
If memory serves, an over-abundance of one of the components in an equilibrium-type reaction would tend to force the equilibrium point in the opposite direction. Hence, this would act as a sort of negative feedback and consequently would somewhat compensate for the initial over-abundance.
Am I understanding this correctly?
Cheers Bill F
[Rob P] - I would recommend you read the OA not OK series (left hand column of the page). Part 7 deals with Le Chatelier's principle.
I do wonder, however, if this series may be slightly revised as some stage because equation 1 is very confusing for most. A naive interpretation could be that increased bicarbonate in the ocean, as a result increased CO2 dissolved in seawater, might be expected to aid biological marine calcification, rather than hinder it.
Bicarbonate is indeed a source of calcification, but it looks like marine calcifiers convert the bicarbonate ion to carbonate in order to form calcium carbonate structures by pumping hydrogen ions out of internal chambers where this calcification takes place. The decrease in the number of hydrogen ions raises the pH of the calcifying fluid considerably and this is what enables the building of the shell or skeleton. There's much more it than that of course, but that's the basic gist.
The concentration of carbonate ions represents an energy gradient upon which calcification must operate. More carbonate ions and less bicarbonate ions make calcification easier, and less carbonate ions and more bicarbonate ions (as in OA) makes shell formation more difficult - the organism has to work harder, pumping more hydrogen ions out of the calcification chamber in order to reach the required level of carbonate saturation.
So, well before carbonate undersaturation is reached (i.e. seawater becomes physically corrosive to calcium carbonate forms), ocean acidification will affect growth rates in many marine organisms.
@ Moderator Comment
Thanks Rob. It's nice to know that there's even more stuff that I don't know anything about, even when I started by knowing so little. ;)
(That's just one of the reasons why I was glad that we only had to do Chemistry in First Year.)
Rob P. @33
I agree the equation may be a bit misleading. The actual calcification step involves carbonate and calcium becoming supersaturating and forming mineral. The organisms promote this by transporting bicarbonate and Ca ions to specific areas of deposition, because those are the ions for which they have transporters, as far as I know. But it is the carbonate concentrations that result when the pH is ratched up that are relevant to mineral formation. High carbonate concentrations are easier to acheive when the pH and the starting concentration of carbonate is greater.
[Rob P] - I sense a blog post/rebuttal in my future, as this aspect has been somewhat glossed over. It's important for readers to be able to close the loop and understand how it all fits together. The calcification process, and how equation one fits into the loop, is one of the missing elements.
Bill @ 33
The problem is that you have a lot of linked reactions, involving different reactants, and phase changes. I had to solve these when studying water chemistry back in the dark ages, but it's been a long time and it's really hard to describe simply how these interact. But for my students I try to focus on four things.
1. Adding CO2 reduces the amount of carbonate in the ocean. This is because the acidity, which is produced when carbonic acid dissociates to form bicarbonate at pH 8 (AO is not OK #8), then combines with preexisting carbonate ions to form more bicarbonate (AO is not OK #7).
2. Decreasing carbonate concentrations make calcium carbonate formation more difficult. This is because calcium carbonate formation is favored when the product of carbonate and calcium ion concentrations is higher (AO not OK #15). Calcifying organisms have a harder time manipulating these concentrations to promote calcification when the background concentrations of these two ions are low.
3. Production of calcium carbonate has the net effect of producing CO2 and increasing acidity (AO is not OK #1). Production of a mole of calcium carbonate by definition removes a mole of bivalent calcium ions from seawater. Change balance must be maintained in seawater to counter this loss. Because univalent bicarbonate ions predominate at pH~8 (AO is not OK #8), approximately two moles of univalent bicarbonate anions must therefore be lost from seawater with each mole of calcium lost. One mole carbon goes to form the 1 mole of calcium carbonate. The other forms H2CO3 that readily dissociates to H2O and CO2, which in turn can exhange with the atmosphere.
(This sequence is behind the equation 1 in the AO is not OK series, I believe. The equation really reflects the net effect of calcification on seawater chemistry rather than what happens exactly at the moment of calcium carbonate precipitation).
4. The opposite reaction (weathering, or dissolution of calcium carbonate) obviously consumes CO2 and produces bicarbonate for the same reasons. Doug brings up the consumption of CO2 by weathering of calcareous deposits on land because it implies that formation of those cliffs must have released CO2. If both the weathering and production of calcium carbonate consumed CO2 you'd have a real problem balancing the equations!
Stephen @ 34
Thanks for this. Your closing bservation... "If both the weathering and production of calcium carbonate consumed CO2 you'd have a real problem balancing the equations!" is certainly intriguing. Sounds a bit like a one-way ticket back to the Cryogenic.
On the other hand, if it was sufficiently exothermic.....
Bill F ;)
Technically, in my # three above I should say...
3. Production of calcium carbonate has the net effect of producing CO2 and reducing alkalinity.
All right already. I'll work on an update.The impact of OA is the change is SW chemistry (specifically the decrease in CO3=). Calcification itself varies for different organisms and is not well studied (compared to say bird egg formation). e.g. consider the case where instead of directly using CO3= a calcifier takes HCO3- from SW or body fluids and then converts to CO3= before forming CaCO3. In such a case the OA caused decrease in SW [CO3=] might appear less important, excpet that actually the decrease in SW [CO3=] increases the dissolution of CaCO3 regardless of how calcification occurs(see posts about common ion and omega). More in update.
[Rob P] - Cheers Doug, that would be rather splendid. And yes, you (and Stephen) make a good point that either manner of calcification is impeded by the decline in carbonate ion abundance. Still, it gives me a opportunity to write about coral calcification & OA.
@18, The trick of Chemistry is called entropy. Which reaction will occur depends on the entropy of the mixture, its temperature but will always strive to get S>0.
Would that help as an answer why some reactions are "preferred" over others?
A late post to this thread, but there is a point related to the original article that is worth noting in case the “Wallace v. Feely” issue arises again.
In addition to spatial and temporal aliasing in the collection of historical data, noted in the original article, one must also be aware that not all pH values are directly comparable. This is because scientists use different “pH scales” (methods of calibration) when determining pH. In a recent review article, Frank Millero noted “Unfortunately, the carbonate constants used to study the CO2 system in the oceans are reported on different pH scales, confusing many trying to understand the carbonate system.” (In case the link doesn’t work, the reference is Treatise on Geochemistry, 2nd edition, Volume 8, Chapter 8.1). Equations 13-16 in the review article show that the same reference seawater can have pH values of 8.32, 8.19, 8,05 and 8.06 on four commonly used pH scales. This range of values is comparable to the change in pH of the ocean over the past century. Historical pH data have been reported on different pH scales, so it is possible that some of the variability in the early 20th century results from the use of different scales, as well as from other problems noted in the original article.
The value of the work by Feely and Sabine is that the results are based on uniform and internally consistent measurements, thereby creating a more robust trend than can be obtained by simply combining all historical data without normalization to a common analytical method.